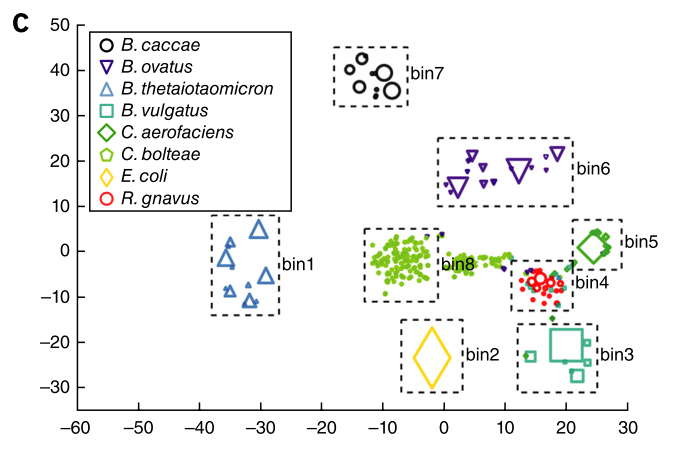
I was rereading a great paper from the Huttenhower group (at Harvard) this week and I was struck by a common theme that it shared with another great paper from the Segre group (at NIH), which I think is a nice little window into how good scientists are approaching the microbiome these days.
The paper I'm thinking about is Hall, et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients (2017) Genome Medicine. The paper is open access so you can feel free to go read it yourself, but my super short summary is: (1) they analyzed the gut microbiome from patients with (and without) IBD and found that a specific clade of Ruminococcus gnavus was enriched in IBD; and then (2) they took the extra step of growing up those bacteria in the lab and sequencing their genomes in order to figure out which specific genes were enriched in IBD.
The basic result is fantastically interesting – they found enriched genes that were associated with oxidative stress, adhesion, iron acquisition, and mucus utilization, all of which make sense in terms of IBD – but I mostly want to talk about the way they figured this out. Namely, they took a combined approach of (1) analyzing the total DNA from stool samples with culture free genome sequencing, and then (2) they isolated and grew R. gnavus strains in culture from those same stool samples so that they could analyze their genomes.

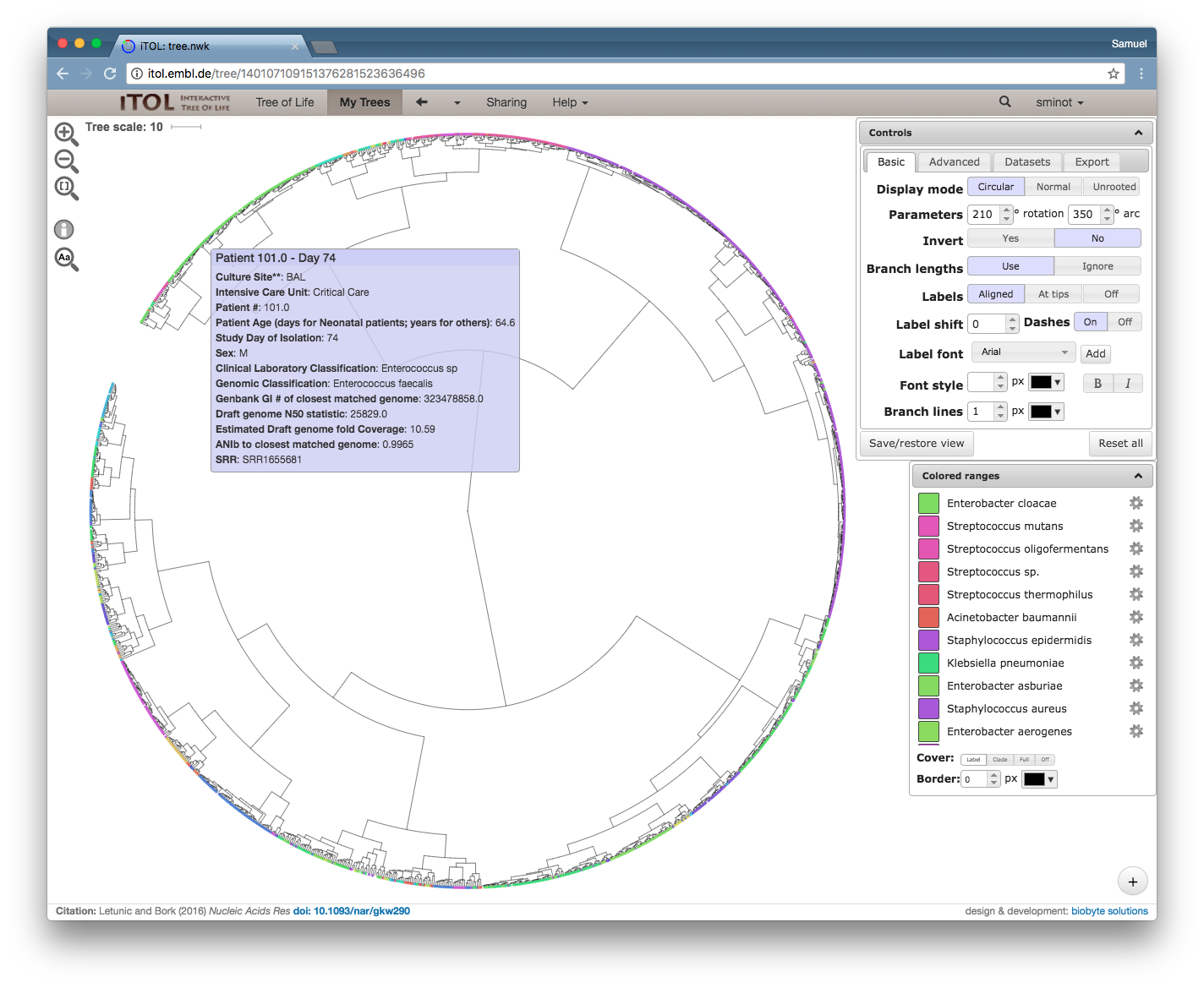
Fig. 3: R. gnavus metagenomic strain phylogeny.
Now, if you cast your mind back to the paper on pediatric atopic dermatitis from Drs. Segre and Kong (Byrd, et al. 2017 Science Translational Medicine) you will remember that they took a very similar approach. They did culture-free sequencing of skin samples, while growing Staph strains from those same skin samples in parallel. With the cultures in hand they were able to sequence the genomes of those strains as well as testing for virulence in a mouse model of dermatitis.
So, why do I think this is worth writing a post about? It helps tell the story of how microbiome research has been developing in recent years. At the start, all we could do was describe how different organisms were in higher and lower abundance in different body sites, disease states, etc. Now that the field has progressed, it is becoming clear that the strain-level differences within a given species may be very important to human health and disease. We know that although people may contain a similar set of common bacterial species, the exact strains in their gut (for example) are different between people and usually stick around for months and years.
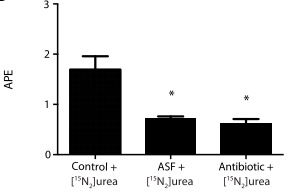
With this increased focus on strain-level diversity, we are coming up against the technological challenges of characterizing those differences between people, and how those differences track with health and disease. The two papers I've mentioned here are not the only ones to take this approach (it's also worth mentioning this great paper on urea metabolism in Crohn's disease from UPenn), which was to neatly interweave the complementary sets of information that can gleaned from culture-free whole-genome shotgun sequencing as well as culture-based strain isolation. Both of those techniques are difficult and they require extremely different sets of skills, so it's great to see collaborations come together to make these studies possible.
With such a short post, I've surely left out some important details from these papers, but I hope that the general reflection and point about the development of microbiome research has been of interest. It's certainly going to stay on my mind for the years to come.