Things have gotten weird. With AI tools available, I find that I can build things I never really could before — web apps and tools in languages I never wanted to touch before. The range of what one person can put together has gotten a lot wider, and I think we’re all still figuring out how to adjust.
Speaking for myself, it used to be that building the thing was the barrier, and now I think the harder question is knowing what to build in the first place — where to actually point our prompts and tokens so that you end up with something useful instead of another half-baked idea.
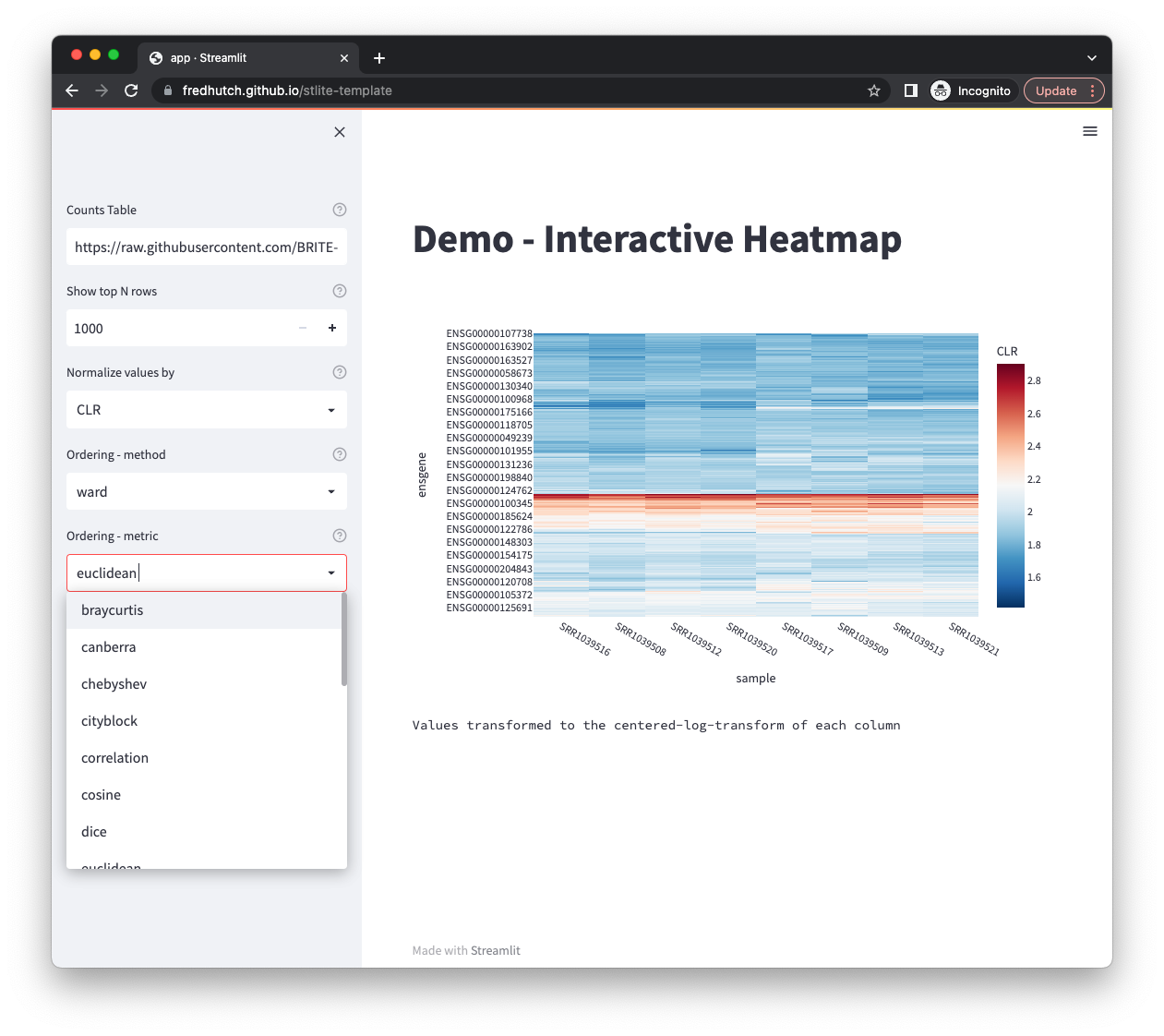
If you're looking for somewhere to explore next, here's my suggestion: build a dashboard with Observable Framework.
I've spent a fair amount of time over the last year telling people about marimo, a reactive notebook for Python that I've become a bit of a broken record about. Observable is sort of the older relative of marimo — same reactive idea, but living in the web browser instead — and for the kind of AI-assisted tinkering I'm describing, it turns out to be a really good fit.
Compared to the languages I’m most comfortable with (Python and R), one of the things I like most about any JavaScript-based tool is just that the code runs in the browser. Everybody already has a browser, which sounds obvious but it's a big deal when you think about how hard it is to get someone else to run your fancy new tool using Python/R/Docker. When I want to share a result with a collaborator, I don't have to walk them through installing anything or ask which version of Python they happen to have — I can send them a link and it opens on their laptop or their phone and that's the end of it. For sharing scientific work, that ends up being really valuable.
What Do I Mean by "Reactive"?
Reactive — a style of programming where the system keeps track of which parts of your code depend on which other parts, and automatically re-runs the parts that need updating when something changes. You describe how things relate to each other, and it works out the order so you don't have to.
If you've spent most of your time in R or Python scripts, this takes a bit of getting used to. In a normal script/notebook the order matters a lot — line 40 needs to run after line 39, or things break. In a reactive setup you stop thinking about order. You just say that this plot depends on this slider, and then when the slider moves the plot updates on its own. You never have to lay out that dependency graph yourself; the runtime infers it while the code is running.
Marimo does the same thing over in Python. For me it's the part that makes both of them feel a bit different from normal coding — the relationships are inferred directly from the code and then you just watch it keep itself in sync.
Where the Idea Came From
I actually first heard about Observable back when I was getting into marimo. The marimo folks talked about having been inspired by it, but at the time it didn't really register. Observable meant writing JavaScript, and I wasn't confident enough to write much of anything in JS, so I sort of filed it away and never looked any closer. It's only more recently, with an assistant to lean on, that going back to the original felt like something I could actually do.
Under the hood they're doing a similar thing with that dependency graph. Where they really differ is in the language you write and in how you get the finished thing out to other people, and that difference matters more than you'd think.
Same Idea, Different Trade-offs
Marimo is Python, which makes it really fun to write if Python is already the language you think in (and if you’ve come up against the pain points of Jupyter). The tricky part is sharing it. Browsers don't run Python on their own, so to put a marimo app in front of someone you either have to run a server for it or bundle up the whole Python runtime and ship it into the browser with something like Pyodide. Both of those work fine, but they each have their own hassles.
Observable is JavaScript, so it just runs in the browser directly, with nothing sitting behind it. An Observable Framework project compiles down to ordinary static files — HTML, CSS, and JavaScript — that you can host just about anywhere, whether that's S3 or GitHub Pages or some cheap static host, with no server to keep running. I've spent an embarrassing amount of my career building static-site tooling to avoid running servers, so this is exactly the kind of thing I tend to get excited about.
So roughly speaking, marimo is easier to write and Observable is easier to share, and which of those matters more really just depends on what you're making and who needs to see it in the end.
Where the Data Comes In
For actual day-to-day work, though, the reactivity on its own isn't really the selling point for me. What I care about is what happens when you hook that reactivity up to a real dataset, and that's where a library called Mosaic comes in.
Mosaic wires your plots up to DuckDB, which can actually run right inside the browser through WebAssembly. So instead of dumping your entire dataset into JavaScript and making the browser chew through all of it, Mosaic hands the filtering and aggregation off to DuckDB and only draws whatever it needs at the moment. In practice that means you can cross-filter a chart sitting on top of a pretty large table and have it stay responsive, which would have been painfully slow in a browser not that long ago. When you drag a selection on one plot, the linked plots re-query the database and update, rather than slogging through a giant array loaded into memory.
Take Home: if you're trying to build a dashboard on top of data that's a bit too big to sit comfortably in a normal web page, the pairing of Observable's reactivity with Mosaic pushing the heavy lifting down to DuckDB is, in my opinion, a really nice way to go about it.
"But I Don't Write JavaScript"
For a lot of the people I talk to — scientists and analysts who mostly live in R or Python — the JavaScript thing has always been where the conversation ends. Observable is JavaScript, they don't write JavaScript, and so they never look any further. One of my closest collaborators tried to convince me to try JavaScript for years, and I always resisted.
And that's the barrier I think has mostly come down. A decent AI coding assistant is quite good at exactly this sort of thing — setting up a small web project, getting a plot working, fixing the layout, or just explaining what some error message actually means. Knowing JavaScript inside and out used to be the entry fee for any of this, and I don't really think it is anymore.
So we're back to that same question of what to actually build. If the building is mostly handled for you now, the more interesting problem is where to aim, and I think Observable is one of the better places to try. The documentation is good, and the whole thing happens to fit how an AI assistant likes to work, so you can lean on it for the JavaScript parts you don't know. You end up writing less code than you'd guess — a lot of it is just Markdown with small bits of JavaScript mixed in — and what you get at the end is a static site you can put anywhere.
Where to Start
If you want to actually try this out, here's roughly where I'd start:
Skim through the Observable Framework docs. The getting-started walkthrough is pretty approachable.
Have a look at one of the Mosaic examples so you can see the big-dataset cross-filtering working, and get a sense of what you're aiming at.
Grab a small dataset of your own and ask your AI assistant to help you build a single interactive chart from it. Just start with one, get it working, and then add another once it does.
The point of a first attempt isn't really to get good at any of this. It's mostly just to get to the moment where you move some control on the page and everything else updates along with it, and you notice that you never actually had to connect those pieces together yourself. I've built a fair number of these by now, and that bit still gets me a little.
—-
If you've been curious about marimo, or about building for the web in general, or you're just tired of exporting static PNGs to drop into slides, I'd really encourage you to give Observable a try. The wall that used to keep the browser out of reach for people who don't write JavaScript is a lot lower now than I think most people realize. Your time and attention are appreciated, as always, dear reader — and if you do end up building something, please be in touch, I'd love to see it.