Amid the holiday rush last month, I was gratified to see a publication describing a new computational method that had been on my mind.
The paper is "Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation", published in Nature Biotechnology on December 11, 2017.
The Concept
Bacteria do something funny to their DNA that you may not have heard of. It's called covalent DNA modification, and it often consists of adding a small methyl group to the DNA molecule itself. This added methyl group (or hydroxyl, or hydroxymethyl, etc.) doesn't interfere with the normal operation of DNA (making RNA, and more DNA). Instead it serves as a marker that differentiates self-DNA from invading DNA (such as from a phage, plasmid, etc.).
For example, one species may methylate the motif ACCGAG (at the bolded base), while another might methylate CTGCAG. If the first species encounters a ACCGAG motif that lacks the appropriate methyl, it treats it as invading DNA and chews it up.
The ongoing arms race of mobile DNA elements has helped maintain a diversity of DNA modifications, such that many different species of bacteria have a unique profile.
The interesting trick here is that we now have a way to read out the methylation patterns in DNA in addition to the sequence. This is most notably available via PacBio sequencing, which typically will generate a smaller number of much longer sequence fragments than other genome sequencing methods. Bringing it all together, the authors of this paper were able to use the methylation patterns from PacBio sequencing to much more accurately reconstruct the microbes present in a set of environmental samples (such as the human microbiome).
The Data
The approach of the authors was to first assemble the PacBio sequences, and then bin those larger genome fragments together based on a common epigenetic signature.
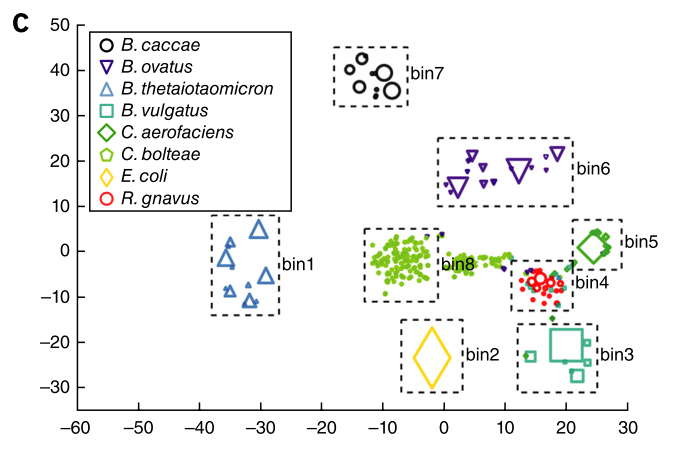
Figure 2c shows the binning of genome fragments in a sample containing a known mixture of bacteria.

Figure 2e shows the binning of genome fragments in a gut microbiome sample containing a mixture of unknown bacteria (and viruses, fungi, etc.).
In general, this seems like a very interesting approach. After my read of the paper, it appears that the bacteria present in the microbiome contain a distinct enough set of epigenetic patterns to enable the deconvolution of many different species and strains. I look forward to seeing how this method stacks up against other binning approaches in the future.
Final note
For those of you interested in phage and mobile genetic elements, I wanted to point out that the authors also explore a topic that has been studied somewhat by others – the linkage of phage to their hosts via epigenetic signatures. The idea here is that it can be computationally difficult to match a phage genome or plasmid with its host. One experimental method that accomplishes this is Hi-C, which takes advantage of the physical proximity of the host genome within an intact cell. In contrast, the PacBio method does not require intact cells, and can be used to link phage or plasmids with their host based on a shared epigenetic signature.
My hope is that this type of data starts to become more widely available. There are clearly a number of computational tools that need to be refined in order to get full use of all this information, but it does seem to hold a good deal of promise.