What is "Genomic Epidemiology"?
The last ten years have seen a number of astonishing advances in our ability to track the spread of outbreaks using something called "genomic epidemiology." The basic concept here is that the pathogens that make us sick (bacteria, viruses, fungi, etc.) each have their own genome, which can be used to trace their ancestry in much the same way that we can use our own genomes to trace our own ancestry.
Just like you can use the human genome to get an idea of how people are related to each other, you can use the microbial genomes to get an idea of how pathogens are related to each other, and therefore how they are spreading between people.
Making sense of tons of data
Without doing a comprehensive survey, I will just say that a number of very smart and capable people have been working on all of the different steps of the process – isolating the pathogen's genome, sequencing it quickly, and comparing those genome sequences to each other. What I wanted to focus on was the final step: taking all of those genomes and getting an idea of what is actually happening. I was inspired here by the NextStrain project, which takes large numbers of genomes for something like Zika and provides an intuitive means for figuring out what's going on.
For my own exploration, I used the data published by Roach, et al. (2015) as they sequenced every single isolate that came through an ICU over the course of a year. I should mention that the laboratory protocols necessary to achieve such a feat are very impressive.
Careful analysis of individual outbreaks involved a number of involved steps to reconstruct bacterial genomes and compare isolates at single-nucleotide resolution. Instead, my goal was to see how quickly I could take the raw data and get an idea of whether there were any clusters that would merit further in-depth investigation (were this an actual surveillance project).
Since all of the raw data was available on SRA, I simply (1) downloaded the raw data with fastq-dump, (2) made MinHash sketches with Finch-rs, (3) calculated pairwise distances between all isolates, (4) performed Ward hierarchical clustering (the neighbor joining tree was too slow), and (5) output a Newick file with the resulting tree structure. I didn't keep track of how long each step took, but start to finish it all took me about 5 hours to finish (including the time spent reading documentation, as well as raw compute).
So here's an example of what that dataset looks like, and here is an interactive link.
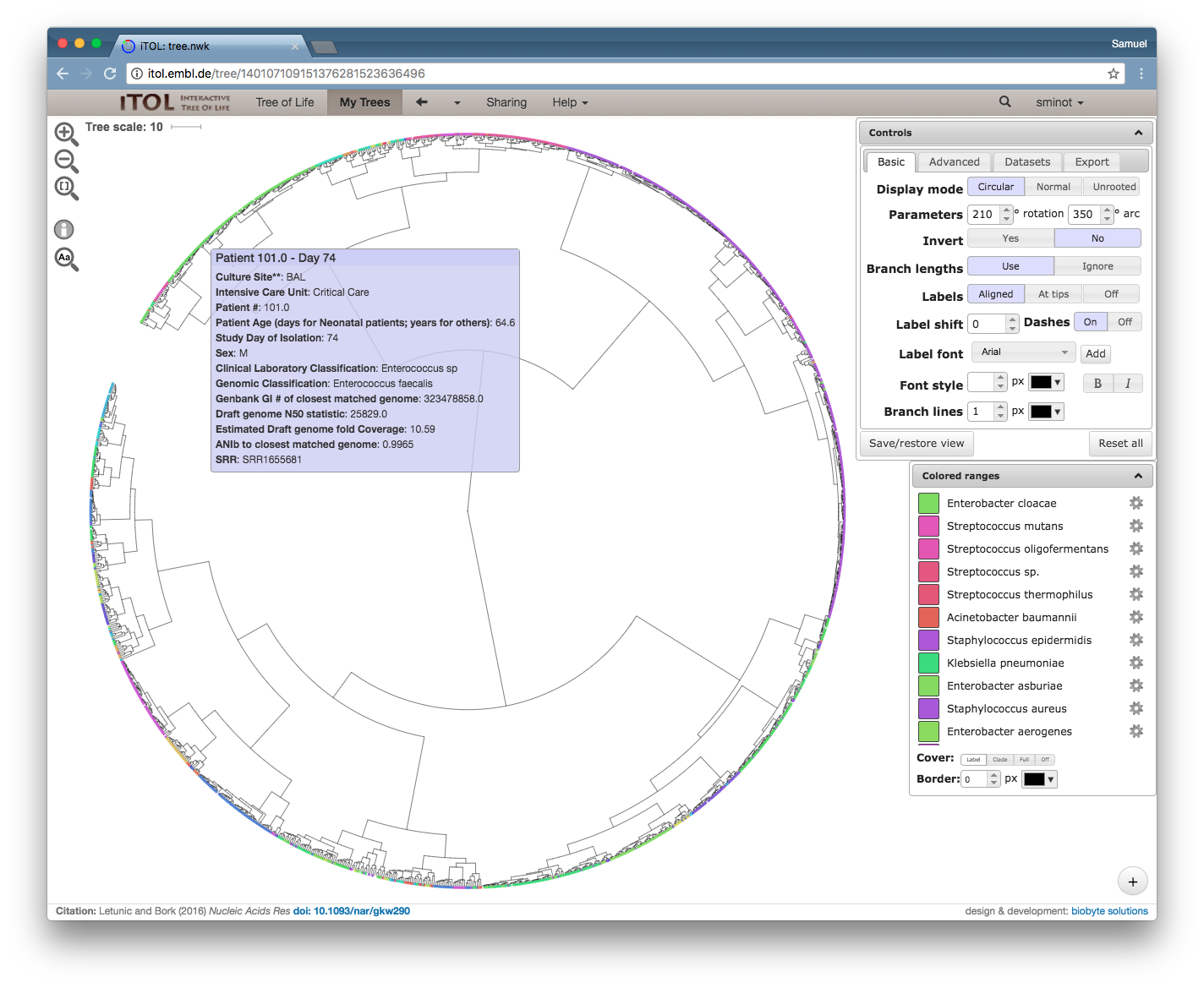
Overall view of 1262 isolates collected over 1 year in an ICU (Roach, et al. 2015)

Zoomed-in view of a clade including Acinetobacter (red), Haemophillus (green), Moraxella (teal), and Neisseria (blue). The rollover text shows metadata for sample 583, which was sampled from the same patient as its closest neighbor in the tree, sample 595.
Disclaimer: None of the clades shown above indicate bona fide outbreak clusters without further quality control, whole genome alignment, and inspection by a qualified professional.
I encourage you to follow the link and explore the tree – it's interesting to see how all of the data comes together. A bunch of different species are included on the same tree, which is one of the nice things about using MinHash sketches to compare genomes (you don't need to align to a common genome). When you roll your mouse over each branch you can see the metadata that the authors published for each isolate.
Take home
So what is the point here? (1) genome sequencing can be used to precisely identify closely-related bacterial isolates, (2) the community (FDA, PHE, CDC, etc.) has taken massive strides to integrate this technology into routine public health surveillance, and (3) relatively simple computational approaches can be used to quickly sift through the data to find groups that merit further inspection.
I also wanted to point out that all of the challenges associated with genome assembly and multiple sequence alignment can be entirely sidestepped by the MinHash approach, which makes it a lot easier to scale these processes up. The analysis methods also don't require a ton of compute – it's actually harder to just keep track of all the data and visualize it in a nice way than it is to compute the pairwise distances.
So, if you're thinking about processing large collections of isolates to find potential outbreak clusters, ask a bioinformatician if MinHash might be right for you.
More Reading:
Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology
Bacterial genome sequencing in clinical microbiology: a pathogen-oriented review
Infection control in the new age of genomic epidemiology
Some MinHash implementations:
Disclosure:
I used to work for and have an interest in One Codex, the group which developed the open source implementation of MinHash (finch-rs) that I used in this little exercise.