Without really having the time to write a full blog post, I want to mention two recent papers that have strongly influenced my understanding of the microbiome.
Niche Theory
The ecological concept of the “niche” is something that is discussed quite often in the field of the microbiome, namely that each bacterial species occupies a niche, and any incoming organism trying to use that same niche will be blocked from establishing itself. The mechanisms and physical factors that cause this “niche exclusion” is probably much more clearly described in the ecological study of plants and animals — in the case of the microbiome I have often wondered just what utility or value this concept really had.
That all changed a few weeks ago with a pair of papers from the Elinav group.
The Papers

Quick, Quick Summary
At the risk of oversimplifying, I’ll try to summarize the two biggest points I took home from these papers.
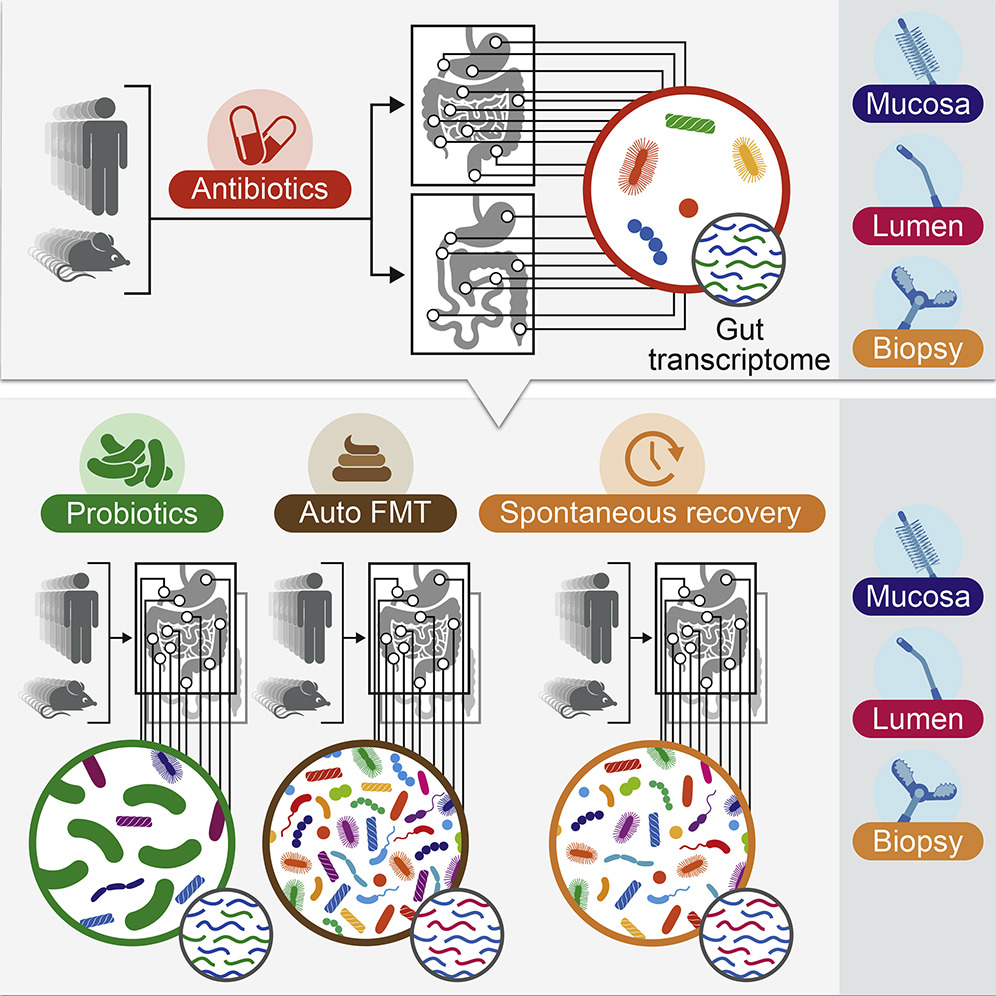
Lowering the abundance and diversity of bacteria in the gut can increase the probability that a new strain of bacteria (from a probiotic) is able to grow and establish itself
The ability of a new bacteria (from a probiotic) to grow and persist in the gut varies widely on a person-by-person basis
Basically, the authors showed quite convincingly that the “niche exclusion” effect does indeed happen in the microbiome, and that the degree of niche exclusion is highly dependent on what microbes are present, as well as a host of other unknown factors.
So many more questions
Like any good study, this raises more questions than it answers. What genetic factors determine whether a new strain of bacteria can grow in the gut? Is it even possible to design a probiotic that can grow in the gut of any human? Are the rules for “niche exclusion” consistent across bacterial species or varied?
As an aside, these studies demonstrate the consistent observation that probiotics generally don’t stick around after you take them. If you have to take a probiotic every day in order to sustain its effect, it’s not a real probiotic.
I invite you to read over these papers and take what you can from them. If I manage to put together a more lengthy or interesting summary, I’ll make sure to post it at some point.